My research interest is centered around the molecular dynamics simulation of biomolecules. In molecular dynamics simulations, molecules are simulated by mechanics (mainly classical mechanics) on computers, and its behaviour is analyzed. Recently I am focusing on to predict the biomolecular characteristics and to computationally (re-)design them.
Molecular dynamics methodologies
One of my research interests I am working for long - though unfortunately it’s not suitable for getting grants in Japan and thus I can’t fully work with - is the development of molecular dynamics methodologies using machine learning. Despite that the core of molecular dynamics is mature, its applications in real-world targets reveal many gaps - the simulation length is orders of magnitude shorter than real world functional events of biomolecules, and the analysis of tera-byte sized trajectory is always hard. I am developing both the calculation methodology and the analysis methodology for molecular dynamics simulation, utilizing my background on bioinformatics and machine learning. One example, Spotting-the-difference paper is a method to analyze the molecular dynamics simulation results. We routinely run two similar simulations with different conditions (e.g., with/without a mutation); this paper is about comparing two (or more) trajectories. In this paper, we pointed out that a simple comparison of average coordinates caused a siginificant artifact, and we proposed to solve the problem with LDA-ITER, a machine learning method developed in the face recognition.
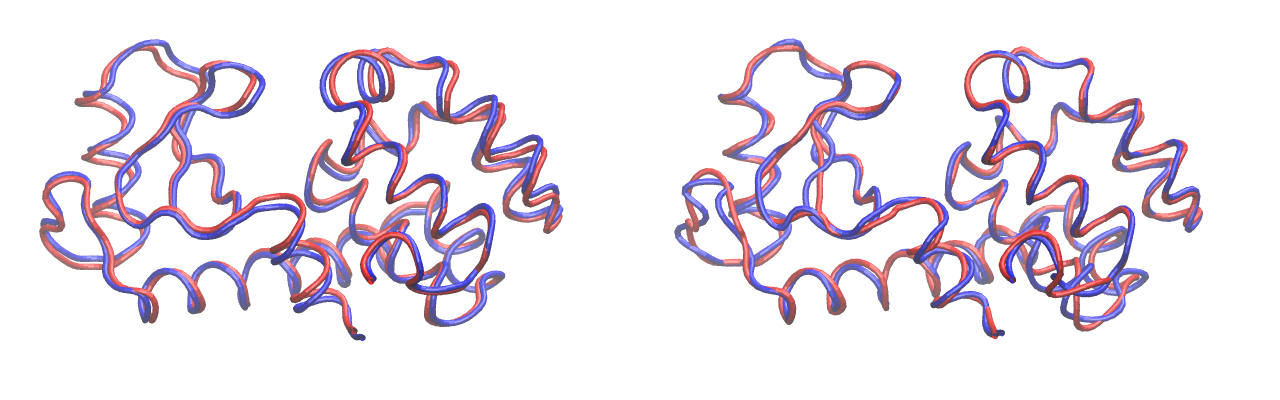 Comparison of generated modes from PCA (left) and LDA-ITER (right). LDA-ITER (right) correctly captures structure differences around the point of mutation.
Comparison of generated modes from PCA (left) and LDA-ITER (right). LDA-ITER (right) correctly captures structure differences around the point of mutation.
You may have thought “Hey, another researcher claiming machine learning and deep learning researches. I’m tired of such claims”; combining machine learning (ML) research to something is such a buzzword now, eh? In this line of research, I am focusing on not using ML just as a tool, but using them to reveal the simulation target’s fundamental, physical structure. You can make an arbitrary “novel” theoretical method replacing parts of previous research to a new one. But it’s not a real progress. I value the math behind the method so that we can “understand” physical meaning. Note I also use ML as just a tool for other lines of work, especially for applications.
- Detecting coupled collective motions in protein by independent subspace analysis
- Spotting the difference in molecular dynamics simulations of biomolecules (Implementation)
Free energy calculations
The term “free energy” is perhaps only well known among those studying physics / physical chemistry; the free energy is, roughly speaking, the quantitative value describing the stability difference. In the context of free-energy calculation of biomolecules, calculating free energies means calculating the equilibrium constants among different states of biomolecules. For example, we can predict the free-energy difference when a drug compound binds to the receptor and when the drug compound leaves the receptor; the result directly indicates the logarithm of the association constant (or the dissociation constant).
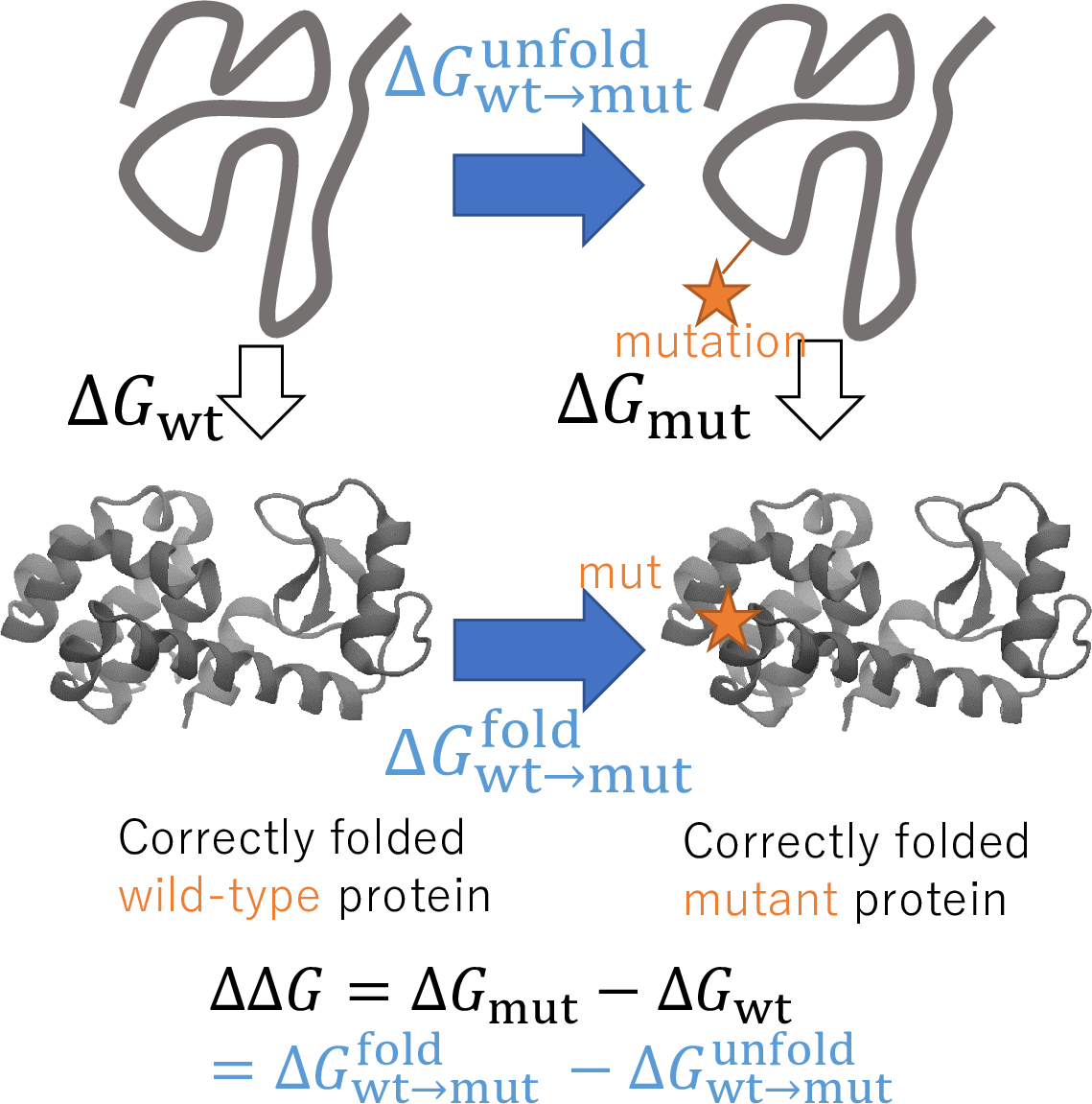
I am recenly focusing protein stability prediction via the free-energy calculation. With the free-energy calculation, we can confidently and thoroughly search the mutation that enhances the stability / function. By combining other pre-screening methods, I can propose mutations with a high “hit” rate; its effectiveness was combat-proven via collaborative works with other research groups (E.g., PCT/JP2022/005051).
- Distribution-function approach to free energy computation
- Free-Energy Calculation of Ribonucleic Inosines and Its Application to Nearest-Neighbor Parameters
- Prediction of ciclesonide binding site on middle-east respiratory syndrome coronavirus nsp15 multimer by molecular dynamics simulations
- A software package is now released as a open-source package as FEP-suite!
Collaborative works with experimental researchers
Computational analysis in collaboration with experimental researchers is another line of my work (recently we are recruiting topics / targets as a part of BINDS(In Japanese)). We needed structure to start the analysis before 2021; now we can start the research with just the sequence thanks to AlphaFold.
One thing I weigh on collaborative works is to find the best approach based on the data and research story, and not to cling to methods we developed in our lab. In fact, in almost all collaborative works, there are no need for fancy methods. 99% of the time, the good old methods that surpassed the test of time suffice; sometimes, the problem is even solved without simulations. I always try to keep a well-known quote in my mind: “If the only tool you have is a hammer, it is tempting to treat everything as if it were a nail”.
- A common allosteric mechanism regulates homeostatic inactivation of auxin and gibberellin
- Histone H3K23-specific acetylation by MORF is coupled to H3K14 acylation
Computational science
Because I had an expertise in programming, I worked on developing efficient algorithms / implementations for scientific computing. Unfortunately I am closing / shrinking this line of works (honestly speaking, it’s because I have been post-doc so long. Software develpments are time-eater and not… ahem, not quite frequently counted as “research works”). I can also modify several open-source program packages (including PLUMED and GROMACS - added new command to PLUMED and modified GROMACS nonbonded kernel as well as integrators.) Some of them are uploaded to GitHub/Bitbucket page.
- ERmod: Fast and versatile computation software for solvation free energy with approximate theory of solutions. (Software page)
- Performance evaluation of the zero-multipole summation method in modern molecular dynamics software
- Large-Scale Parallel Implementation of Hartree-Fock Exchange Energy on Real-Space Grids Using 3D-Parallel Fast Fourier Transform (Code)