生体分子の分子動力学法による解析を核として研究を行っています。分子動力学法は分子の運動をコンピューター上で力学に従ってシミュレーションし、分子がどのように振る舞うかを解析することで、分子のもつ性質を調べる手法です。特に最近では、生体分子の機能・性能を予測し、生体分子の改変・創製に使う方向性を模索しています。
分子動力学法の手法開発
長年力を入れている(あまり予算取り向きでは無いので、熱意とお仕事としての比重が一致しないのですが)のは機械学習アルゴリズムを応用した分子動力学法の手法開発です。分子動力学法は手法として確立して日が長いですが、実際に現実のターゲットと向き合っていると力不足を感じるところや、解析の難しさなどがあります。分子動力学法の持つポテンシャルをもっと引き出すために、計算手法・解析手法の双方の開発を行っています。自分はバイオインフォマティクス・機械学習のバックグラウンドを持つため、これらの分野の知識を積極的に援用する形で手法開発に取り組んでいます。たとえば間違い探し論文は分子動力学法の計算結果の解析手法として提案したものです。我々は異なる条件で2通りのシミュレーション(例: ある残基への変異の有り無し)を行い、比較することを頻繁に行います。本研究はこのトラジェクトリの比較の際に、通例使われる平均構造の比較がアーティファクトをもたらすことを指摘し、顔認識の分野で登場した機械学習手法LDA-ITERを用いることでこれを解決しています。
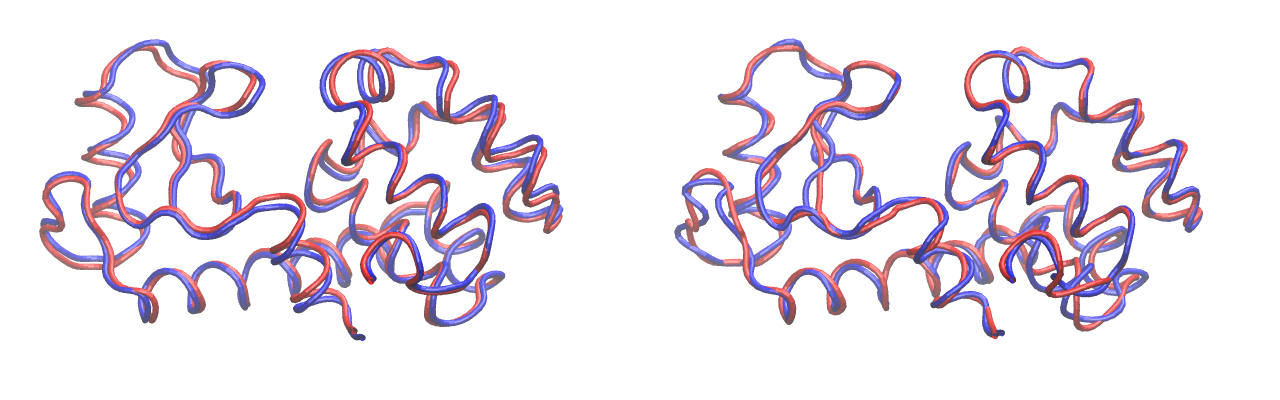 Comparison of generated modes from PCA (left) and LDA-ITER (right). LDA-ITER (right) correctly captures structure differences around the point of mutation.
Comparison of generated modes from PCA (left) and LDA-ITER (right). LDA-ITER (right) correctly captures structure differences around the point of mutation.
といっても、ここ数年間はどこの先生も機械学習をキーワードに入れているので、「まーたこいつもかよ」と懐疑的な目で見られる方も多いかなと思います。自分が特に重視しているラインは、「基礎研究なのだから、機械学習をただのツールとして使うのでは無く、ちゃんと数式に立ち返って何をやっているのか考える」です。物理学的な意味が明らかである(手法を使うことでより良く「わかった」と言える)ような研究を目指しています(ちょっと古いですが、総説を見ると私が目指すところの雰囲気が分かるかもしれません)。一方で、応用研究では機械学習を完全にツールと割り切って使っています。
- Detecting coupled collective motions in protein by independent subspace analysis
- Spotting the difference in molecular dynamics simulations of biomolecules (実装)
自由エネルギー計算
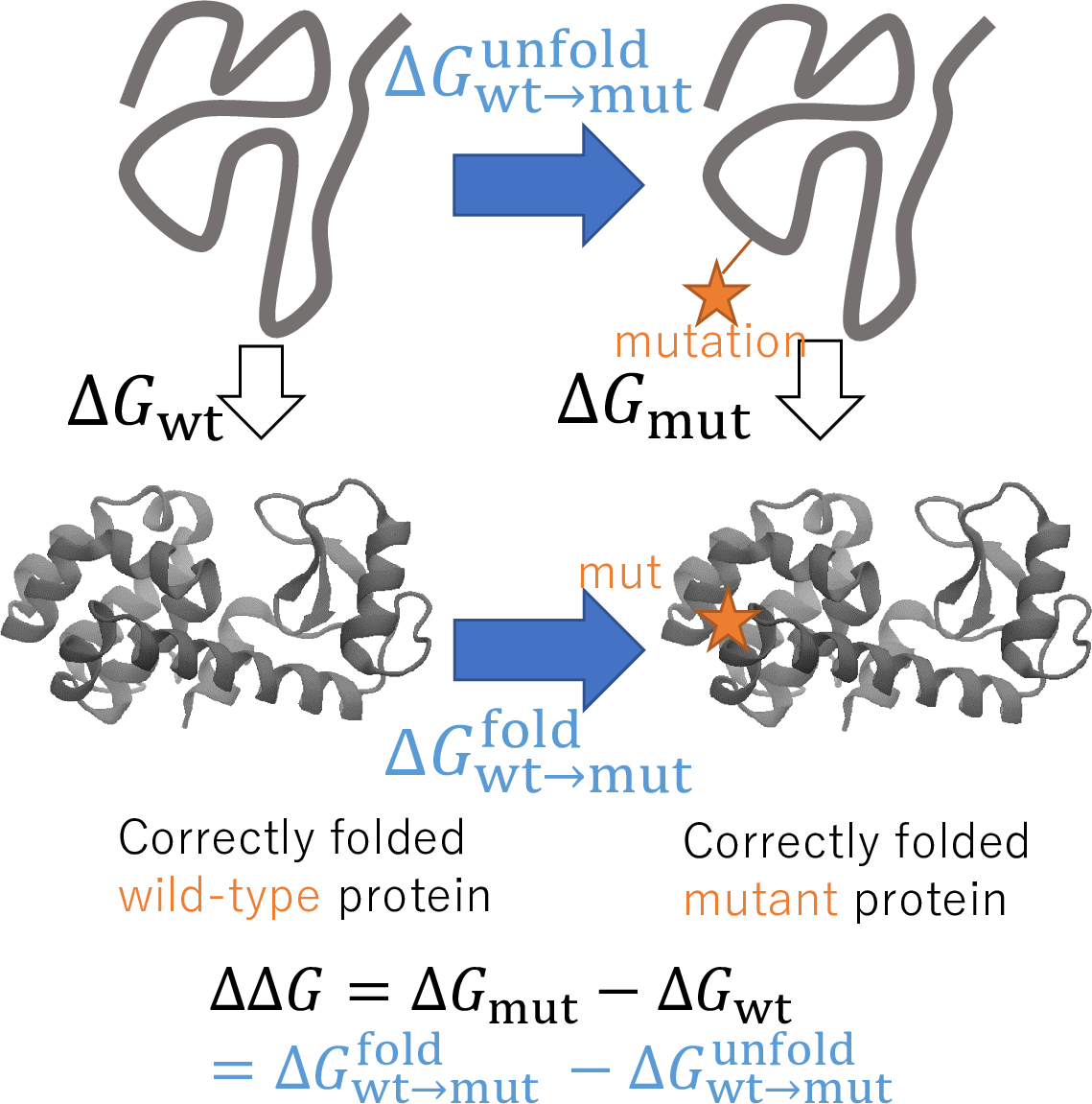
自由エネルギーというと、物理・物理化学屋さん以外には馴染みが無い単語では無いかと思いますが、ざっくりと「安定性の差」を表す定量的な値に相当します。 生体分子シミュレーションの文脈では、自由エネルギーを計算することは、生体分子の様々な状態の間の平衡定数を決めることと同値です。たとえば、薬剤分子が結合したときと、薬剤分子が離れたとき、その安定性の差を計算すると薬剤分子の結合定数(or 乖離定数)が予測できます。
特に、最近では計算機によるタンパク質の安定性予測に注力しています。自由エネルギー差を求めることでタンパク質の安定性・機能を向上させる変異を計算によって網羅的に探索することができる手法を開発しています。本手法は他のプレスクリーニング法と組み合わせることで高いヒット率で安定化変異を提案でき、複数の研究グループとの共同研究で非常に良い成果を上げています(公開になったものといえばたとえばPCT/JP2022/005051)。
- Distribution-function approach to free energy computation
- Free-Energy Calculation of Ribonucleic Inosines and Its Application to Nearest-Neighbor Parameters
- Prediction of ciclesonide binding site on middle-east respiratory syndrome coronavirus nsp15 multimer by molecular dynamics simulations
- FEP周りの自作ソフトをパッケージングして FEP-suite として公開しました。
共同研究系
実験グループとの共同研究で様々な計算による解析を行っています(最近は主にBINDSの一環として受けることが多いです)。2020年までは(最低でもホモログの)結晶構造が無いと研究を開始できなかったのですが、最近ではAlphaFoldによって構造予測の精度が劇的に向上したため、配列だけあればなにかしらできるようになりました。
共同研究で重視していることは、「自分たちで開発した手法にこだわらず、研究のストーリーを踏まえて最適なアプローチを探す」ということです。というより、ほとんどの共同研究案件には新たな手法は不要で、年月の厚みに耐えてきた手法だけで片が付きます(時にはシミュレーションすら不要なこともあります)。持つ道具がハンマーしかなければ、すべてが釘に見える、を心に銘じています。
- A common allosteric mechanism regulates homeostatic inactivation of auxin and gibberellin
- Histone H3K23-specific acetylation by MORF is coupled to H3K14 acylation
Computational science / 計算科学的な仕事
元々計算機系に強かったので、効率の良い計算科学用アルゴリズムの考案、ソフトウェアの整備などをやっていました。 最近はこの手の仕事は縮小気味です(身も蓋もない話をするとソフトウェア系は時間を食うのに業績にならず、またuntenuredな期間が長かったので……)。 GROMACSやPLUMEDへの機能追加ができます。GROMACSのnonbonded kernelや積分子の改造を行った経験もあります。他、上で挙げたネタをソフトウェアにして整備したりしています(いくつかはbitbucketやgithubで公開しています)
- ERmod: Fast and versatile computation software for solvation free energy with approximate theory of solutions. (Software page)
- Performance evaluation of the zero-multipole summation method in modern molecular dynamics software
- Large-Scale Parallel Implementation of Hartree-Fock Exchange Energy on Real-Space Grids Using 3D-Parallel Fast Fourier Transform (Code)